Welcome into the fold
Find out more at www.proteinqure.com
Protein folding is a “holy grail” problem in the field of biology. Chemists, biologists, and physicists have pursued it for decades, uncovering astonishing complexity. Solving it would be grounds for a Nobel prize. Molecular modelling and simulations have been fundamental tools for understanding it. These simulations account for a huge fraction of the world’s supercomputing usage, and have driven the engineering of entirely new computing architectures¹. In a subsequent article we will discuss how quantum computing may result in major breakthroughs in this area, but today we will focus on describing this problem at a high level.
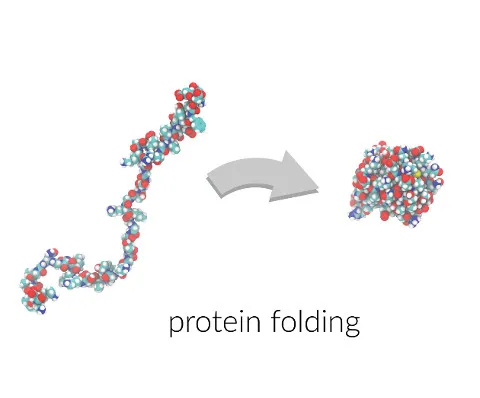
Proteins are large bio-molecules made up of one or more chains of amino acids. Proteins differ from each other in terms of the sequence of those amino acids and length of these chains². This unique sequence determines the protein fold. Proteins fold into a complex three-dimensional shape (or structure); and that shape determines its function and activity in the body. These complicated molecules participate in almost every process within cells, and knowing their structure unlocks a massive potential for understanding life on earth.

The protein folding problem was first described 50+ years ago. It is best articulated by Ken Dill one of the founder’s of the field³:
(i) What is the physical code by which an amino acid sequence dictates a protein’s native structure? (ii) How can proteins fold so fast? (iii) Can we devise a computer algorithm to predict protein structures from their sequences?
The scientific community has made significant progress on part (i), but not as much on the other aspects. ProteinQure is on the forefront of solving the third question; developing new methods to determine the structure of proteins given only its amino acid sequence.
. . .
What happens when we get it
If solved, the protein folding problem unlocks many of the diseases facing humanity. Scientists would have methods to help develop treatments for diseases directly related to protein folding like Alzheimer’s and Creutzfeldt-Jakob disease. Thousands of proteins associated with diseases could have their unknown structures solved with these computational methods. We can use the rules of folding to design new protein structures which encode new and exciting functionality. The physical shape of a molecule, big or small, governs what impact it can have in the body, which makes proteins a powerful class of therapeutics.
Once we start to understand the folding of proteins this allows us to move towards designing them. The capability to generate sequences of amino acids that will fold into a desired 3D structure is a type of “reverse protein folding”. However, designed proteins also have diverse applications:
- New therapeutics (that’s us!)
- Agriculture such as insecticidal proteins or frost protective coatings⁴
- Tissue regeneration through self-assembling proteins⁵
- Cosmetics and supplements for improved health (strength or anti-aging)⁶
- Biomaterials for textiles and materials (spider silk is all protein!)⁷
So solving the protein folding problem doesn’t just mean curing diseases. It means helping solve world hunger, super-humans and cool new jackets. No big deal.
The physics and biology of protein folding
Proteins are not stable in their unfolded, or “fully extended”, shape. Proteins spontaneously fold, inside and outside of the body, based on interactions between amino acids and with its environment. Some amino acids may cause the protein chain to clump up onto itself, shielding the core of the protein from surrounding water molecules, while other amino acids may adopt no preferred structure and perpetually flop around in search for its interaction partner. This process is driven by energy minimization. You can imagine the process of protein folding as a hiking expedition across a scenic field of rolling hills for the lowest, deepest valley.
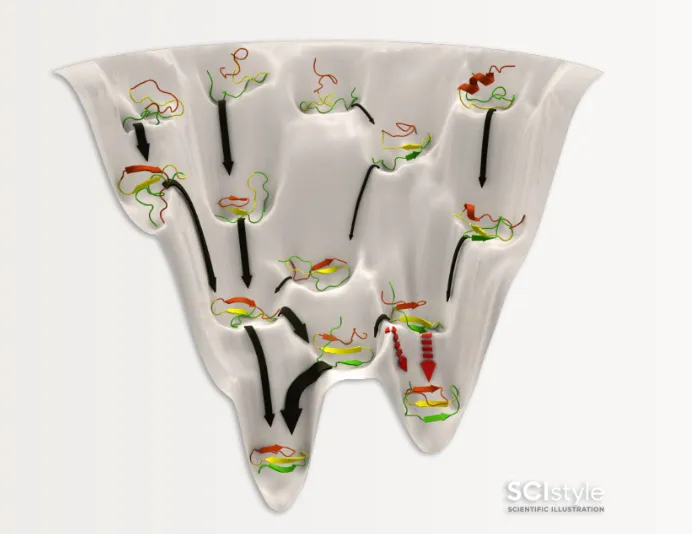
Here, the tops of the hills describe protein folds that are very unlikely to occur, and the valleys describe states that the protein is drawn towards, the native state.
A great puzzle of protein folding is how the body was able to find the folded state so quickly. Simply checking all the possible folds to find the lowest energy one is almost impossible. For a given protein of N amino acids, there are approximately 3^N possible conformations (folds) and this quickly becomes intractable for proteins of meaningful size (relevant proteins in our body can easily approach 100s of amino acids)⁸. The energy landscape itself is both high-dimensional and extremely rugged. Trying to predict the shape of the landscape remains difficult for most methods. So how do scientists approach the immense complexity of this problem when using a computer?
. . .
Getting computers to do biology
There are two main approaches to use computers to produce structures from sequences. The first is to simulate the underlying physics, as described above, and the second is to use knowlege-based approaches which build on pre-existing knowledge about protein structures. Both approaches have strengths and weaknesses, and they aren’t necessarily mutually exclusive.
The first approach is to use computers to simulate the physics of protein folding. Protein models incorporate the basic laws of physics and simulate the interactions of atoms over time. Physical simulations have been used to fold a range of fast-folding proteins starting from the unfolded state⁹. Unfortunately, many interesting proteins fold on the timescale of seconds, something that requires prohibitively large computing resources to simulate, even using the fastest and largest supercomputers on earth¹⁰. One strategy to fold proteins faster is to incorporate shortcuts or assumptions to help us make our algorithms more efficient.
The second approach is to use knowledge-based methods; leveraging energy functions, databases, and machine learning. These techniques reign supreme in protein structure prediction competitions and require significantly less computing power. These methods are most accurate when similar proteins or protein fragments can be found in structure databases. For example, a specific sequence of amino acids may be known to give rise to an helix structure, so it would be possible to build a new protein by integrating this fragment into the context of a larger protein. These methods have led to groundbreaking discoveries in novel protein design¹¹.

The problem with the latter approach is data availability. There are 150,000 known structures in the protein databank and we add about 10,000 every year. If we just consider 50 amino acid length proteins, there are over 10⁶⁵ possible proteins. Many potentially useful proteins are next to impossible to discover given the limited data we have available. Physical simulations are a promising method for exploring protein structures outside of known databases due to their generality. Moving forward, we’re excited about the integration of both methods to drive protein design, as both have the potential to be accelerated by optimization problems solved on quantum computers.
. . .
ProteinQure’s thesis is that by solving the protein folding problem we will be able to unlock applications that create massive impact in the world. Whether you are excited by the fundamental biological questions, earliest practical applications of quantum computing or latest in reinforcement learning; we welcome you into the fold.
Written with contributions from Christopher Ing
[1] https://www.nature.com/articles/nbt0100_8d
[2] https://en.wikipedia.org/wiki/Protein
[3] http://science.sciencemag.org/content/338/6110/1042.full
[4] http://www.proteininnovations.ca/
[5] https://pubs.acs.org/doi/abs/10.1021/cr300131h
[6] https://en.wikipedia.org/wiki/Klotho_(biology)
[8] https://en.wikipedia.org/wiki/Levinthal%27s_paradox
[9] http://science.sciencemag.org/content/334/6055/517.full
[10] https://en.wikipedia.org/wiki/Anton_(computer) , https://en.wikipedia.org/wiki/Folding@home
[11] https://www.nytimes.com/2017/12/26/science/protein-design-david-baker.html